Phylogenetic Analysis of Pathogenic Candida Species Isolated from Iraqi Patients
Abstract
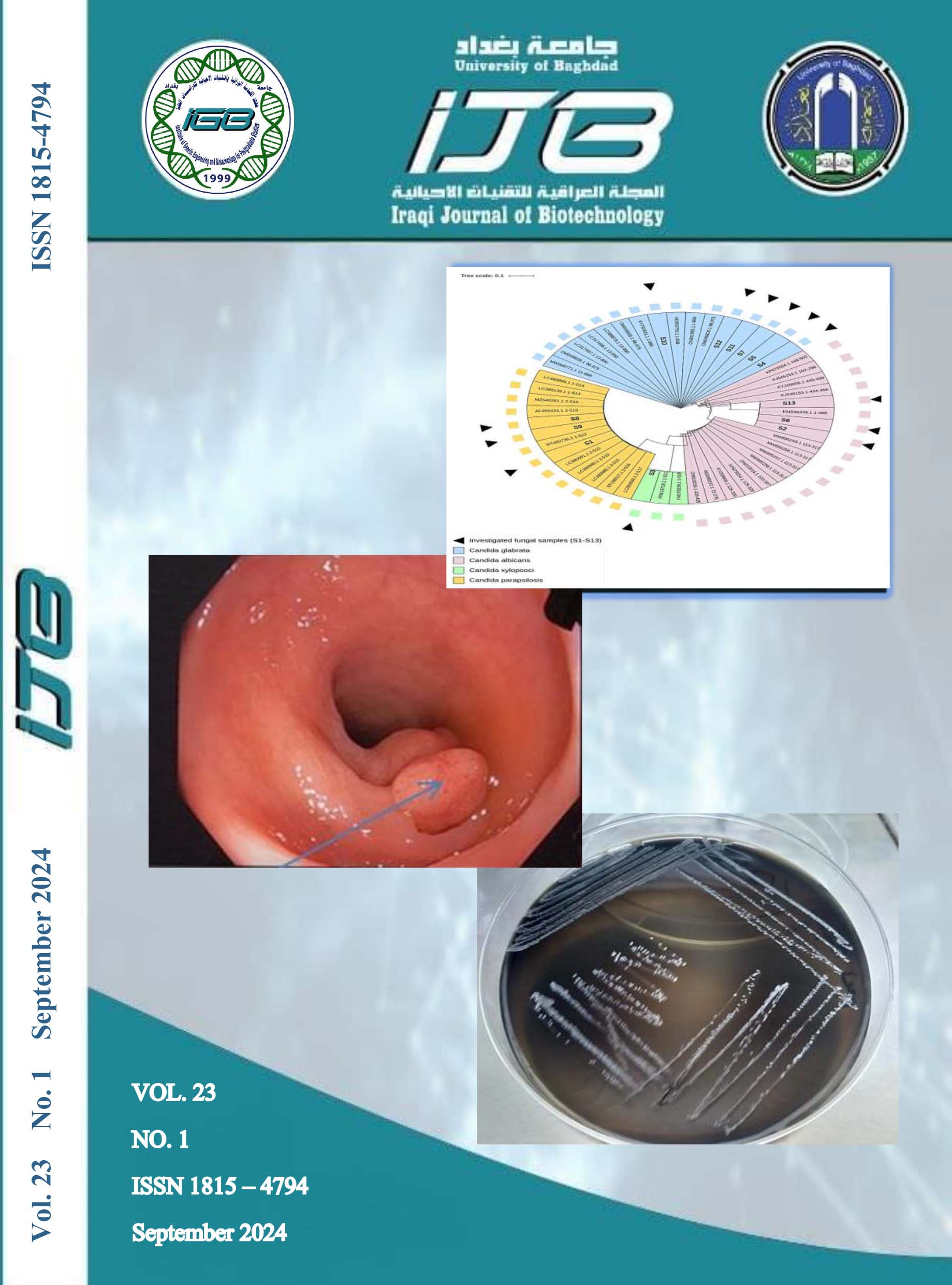
Pathogenic species of Candida have extensively contributed to the raising mortality, morbidity and globally increase medical prices of healthcare-related infectious diseases. The current study was focused on isolation and identification of Candida spp. from different clinical specimens and their molecular phylogenetic analyses. The study was included clinical specimens (100) from different parts of the patient’s body, such as (ear, mouth. vagina, skin, blood and urine) who attended to Gazi Al Hariri hospital, Baghdad teaching hospital in Medical City and Al-Yarmouk teaching hospital. The isolation of Candida spp. using selective media and vitek system. Then, a full inclusive tree, including the observed variant, was built by the neighbour-joining method and visualized as a circular cladogram using the iTOL suit. The findings of isolation and identification of Candida spp. in studied samples were recorded that the highest isolate was C. parapsilosis (27.7%) followed by C glabrata (22.2%), while the lowest isolates were C. tropicalis and C. krusei (5.50%) each one, Candida spp. also identified by ITS which results showed the presence of sizes ranging 550 - 625 bp depending on the difference in the lengths of the region (ITS 1 and ITS4) for yeast species, so they produce different sizes of DNA pieces. Sequencing reactions showed the accurate identity of the investigated samples and revealed that S1, S8, and S9 were homologous to C. parapsilosis, S2, S6, and S13 were similar to C. albicans, S3 was similar to C. xylopsoci while S4, S5, S7, S10, S11, and S12 were similar to C. glabrata (The presence of a total of 11 nucleic acid variants compared with the referring sequences of the Candida sequences was demonstrated. The identified variants were one deletion (g.492-494Adel) in S1, two substitutions (g.168A>G and g.233C>A) in S13, one substitution (g.83A>C) in S3, one substitution (g.116T>C) in S11, and four substitutions and one deletion (g.339C>A, g.353G>A, g.618G>A, g.356-359GGdel) in S4, S5, S7, S11, S12, one substitution in S7 (g.345C>G), one substitution (g.805T>C) in S4, S5, and S7. Meanwhile, the rest of the samples had shown a complete homology with the corresponding sequences and did not exhibit any detectable nucleic acid variations in comparison with the same reference sequences. This study suggests possible employment for these ribosomal amplicons to discriminate between the phylogenetic diversity among the other implemented tools.